
Les réglementations pharmaceutiques sont le fondement de l’industrie pharmaceutique, garantissant que tous les médicaments répondent aux plus hautes normes de sécurité, d’efficacité et de qualité. Ces réglementations sont cruciales pour les entreprises pharmaceutiques, alors qu’elles naviguent dans le processus complexe de mise sur le marché de nouveaux médicaments tout en maintenant la conformité aux normes mondiales. Ce guide vise à fournir une compréhension approfondie des réglementations pharmaceutiques, offrant des aperçus des divers cadres qui régissent le développement, l’approbation et la distribution des produits pharmaceutiques. Que vous soyez novice ou expert de l’industrie, ce guide complet vous aidera à rester informé et conforme dans un paysage réglementaire en constante évolution.
Comprendre les réglementations pharmaceutiques
Que sont les réglementations pharmaceutiques ?
Les réglementations pharmaceutiques sont un ensemble de lois, directives et normes qui régissent tous les aspects de l’industrie pharmaceutique, du développement des médicaments à leur commercialisation et leur vente. Ces réglementations sont conçues pour garantir que les produits pharmaceutiques sont sûrs, efficaces et de haute qualité. Les organismes de réglementation tels que la Food and Drug Administration (FDA) aux États-Unis et l’Agence européenne des médicaments (EMA) en Europe jouent un rôle important dans l’application de ces réglementations, veillant à ce que tous les médicaments se conforment aux exigences strictes nécessaires pour protéger la santé publique.
Le rôle des autorités de réglementation des médicaments
Les autorités de réglementation des médicaments sont responsables de la supervision du développement, de l’approbation et de la surveillance des produits pharmaceutiques. Ces autorités, y compris la FDA et l’EMA, s’assurent que tous les médicaments répondent aux normes nécessaires de sécurité et d’efficacité avant d’être approuvés pour un usage public. Elles jouent également un rôle crucial dans la surveillance post-commercialisation, surveillant la sécurité des médicaments par le biais de rapports d’événements indésirables et d’examens périodiques.
Impact sur l’industrie pharmaceutique
L’industrie pharmaceutique est l’un des secteurs les plus réglementés au monde. Les réglementations pharmaceutiques ont un impact sur chaque étape du processus de développement des médicaments, de la recherche et du développement initiaux aux essais cliniques, en passant par la fabrication et la commercialisation. La conformité à ces réglementations est essentielle pour les entreprises pharmaceutiques afin d’éviter les problèmes juridiques, d’assurer la sécurité des médicaments et de maintenir la confiance du public.
Aspects clés de la conformité réglementaire pharmaceutique
Bonnes pratiques de fabrication actuelles (BPF)
L’un des composants les plus critiques de la réglementation pharmaceutique est l’adhésion aux directives actuelles des Bonnes Pratiques de Fabrication (BPF). Les BPF garantissent que les fabricants de médicaments suivent des normes rigoureuses pour la qualité des produits, notamment en maintenant des installations propres, en formant correctement les employés et en documentant minutieusement les processus de fabrication. La conformité aux BPF est obligatoire pour tous les fabricants pharmaceutiques, car elle aide à prévenir la contamination, assure la cohérence des produits et garantit que tous les médicaments sont sûrs pour l’utilisation humaine.
Assurer la sécurité et l’efficacité des médicaments
Assurer la sécurité des médicaments est une priorité absolue pour les autorités réglementaires. Cela implique des tests rigoureux pendant le processus de développement du médicament, y compris des études précliniques et des essais cliniques, pour évaluer la sécurité et l’efficacité des nouveaux médicaments. Les agences de réglementation comme la FDA exigent des données exhaustives pour démontrer qu’un nouveau médicament est sûr et efficace avant qu’il puisse être approuvé pour un usage public. La surveillance post-commercialisation joue également un rôle crucial pour assurer la sécurité continue des médicaments, permettant la détection d’événements indésirables et la mise en œuvre des mesures de sécurité nécessaires.
Conformité aux normes internationales
Les entreprises pharmaceutiques qui opèrent à l’échelle mondiale doivent se conformer à une gamme de normes internationales. Le Conseil international d’harmonisation des exigences techniques pour l’enregistrement des médicaments à usage humain (ICH) joue un rôle clé dans l’harmonisation des normes réglementaires dans différentes régions. La conformité à ces normes garantit que les produits pharmaceutiques répondent aux critères nécessaires de qualité, de sécurité et d’efficacité sur de multiples marchés, facilitant ainsi les ventes et la distribution globales de médicaments.
Orientation et soutien réglementaires
Aperçu des services réglementaires
Naviguer dans les complexités des réglementations pharmaceutiques nécessite un soutien réglementaire expert. Les entreprises pharmaceutiques s’appuient sur des services spécialisés pour assurer la conformité tout au long des processus de développement et d’enregistrement des médicaments. Ces services comprennent la surveillance continue, l’évaluation des informations réglementaires, la rédaction de rapports scientifiques, l’évaluation des risques et la compilation de dossiers d’enregistrement selon les normes les plus élevées. Un soutien réglementaire efficace est essentiel pour garantir que toutes les soumissions, y compris les demandes de nouveaux médicaments, les variations et les renouvellements, répondent aux critères réglementaires requis.
Surveillance continue et conformité
La surveillance continue est une composante vitale de la conformité réglementaire. Elle implique l’examen et la mise à jour réguliers de la documentation, l’évaluation des risques réglementaires et la garantie que toutes les opérations de fabrication adhèrent aux normes BPF. En maintenant une surveillance continue, les entreprises pharmaceutiques peuvent traiter de manière proactive les problèmes potentiels de conformité, minimisant ainsi le risque de retards réglementaires et assurant un accès rapide au marché pour leurs produits.
Évaluation des risques et rapports scientifiques
L’évaluation des risques est un aspect crucial de la conformité réglementaire, particulièrement dans des domaines tels que les nitrosamines et les impuretés élémentaires. Les entreprises pharmaceutiques doivent effectuer des évaluations des risques approfondies pour identifier les dangers potentiels et mettre en œuvre des mesures pour les atténuer. Les rapports scientifiques sont également essentiels, car ils fournissent les données nécessaires pour soutenir les soumissions réglementaires et démontrer la conformité aux normes de sécurité et de qualité.
Réglementations et directives de la FDA
Aperçu des directives de la FDA
La Food and Drug Administration (FDA) est l’organisme de réglementation principal pour les produits pharmaceutiques aux États-Unis. Les directives de la FDA couvrent tous les aspects du développement des médicaments, de la recherche préclinique à la surveillance post-commercialisation. La conformité à ces directives est obligatoire pour toutes les entreprises pharmaceutiques qui souhaitent commercialiser leurs produits aux États-Unis. Les directives de la FDA évoluent constamment, il est donc essentiel pour les entreprises pharmaceutiques de rester à jour avec les derniers changements réglementaires.
La loi de modernisation de la Food and Drug Administration
La loi de modernisation de la Food and Drug Administration (FDAMA) est une législation importante qui a introduit plusieurs changements dans le processus d’approbation des médicaments. La FDAMA visait à rationaliser le processus d’approbation des nouveaux médicaments, le rendant plus rapide et plus efficace. Elle a également introduit de nouvelles exigences pour la surveillance post-commercialisation et accru l’autorité de la FDA pour faire respecter la conformité aux directives BPF.
Conformité à la loi sur les amendements à l’administration des médicaments
La loi sur les amendements à l’administration des médicaments (DAAA) a introduit des exigences supplémentaires pour la sécurité des médicaments et la surveillance post-commercialisation. La DAAA a donné à la FDA de nouveaux outils pour surveiller la sécurité des médicaments, y compris l’autorité d’exiger des stratégies d’évaluation et d’atténuation des risques (REMS) pour certains médicaments. La conformité à la DAAA est essentielle pour garantir que les médicaments restent sûrs et efficaces tout au long de leur cycle de vie.
Réglementations et directives de l’EMA
Aperçu de l’Agence européenne des médicaments (EMA)
L’Agence européenne des médicaments (EMA) est l’autorité réglementaire centrale responsable de l’évaluation, de la supervision et de la surveillance de la sécurité des médicaments au sein de l’Union européenne. Établie en 1995, l’EMA joue un rôle crucial en veillant à ce que tous les produits pharmaceutiques disponibles dans l’UE répondent aux normes les plus élevées de sécurité, d’efficacité et de qualité. L’agence opère à travers un processus d’approbation des médicaments centralisé, permettant aux entreprises pharmaceutiques de soumettre une seule demande d’autorisation de mise sur le marché qui, si approuvée, est valable dans tous les États membres de l’UE. Ce processus est particulièrement important pour les médicaments innovants et à fort impact, assurant un accès rationalisé aux nouveaux traitements pour les patients à travers l’Europe. Le Comité des médicaments à usage humain (CHMP) de l’EMA effectue des évaluations scientifiques rigoureuses pour évaluer les avantages et les risques des nouveaux médicaments, guidant ultimement la décision sur l’approbation d’un produit pour le marché européen.
Directives de l’EMA et exigences de conformité
Au-delà des approbations de médicaments, l’EMA fournit des directives et un soutien réglementaire étendus pour aider les entreprises pharmaceutiques à se conformer aux réglementations de l’UE tout au long du cycle de vie d’un médicament. Cela inclut la surveillance post-commercialisation à travers un système robuste de pharmacovigilance qui surveille la sécurité des médicaments une fois qu’ils sont disponibles pour le public. L’EMA collabore également avec les autorités réglementaires nationales et les organismes internationaux comme la FDA pour harmoniser les normes et partager des informations critiques sur la sécurité. Pour les entreprises pharmaceutiques, la conformité aux réglementations de l’EMA est essentielle non seulement pour obtenir l’accès au marché, mais aussi pour maintenir la sécurité et l’efficacité continues de leurs produits sur le marché européen.
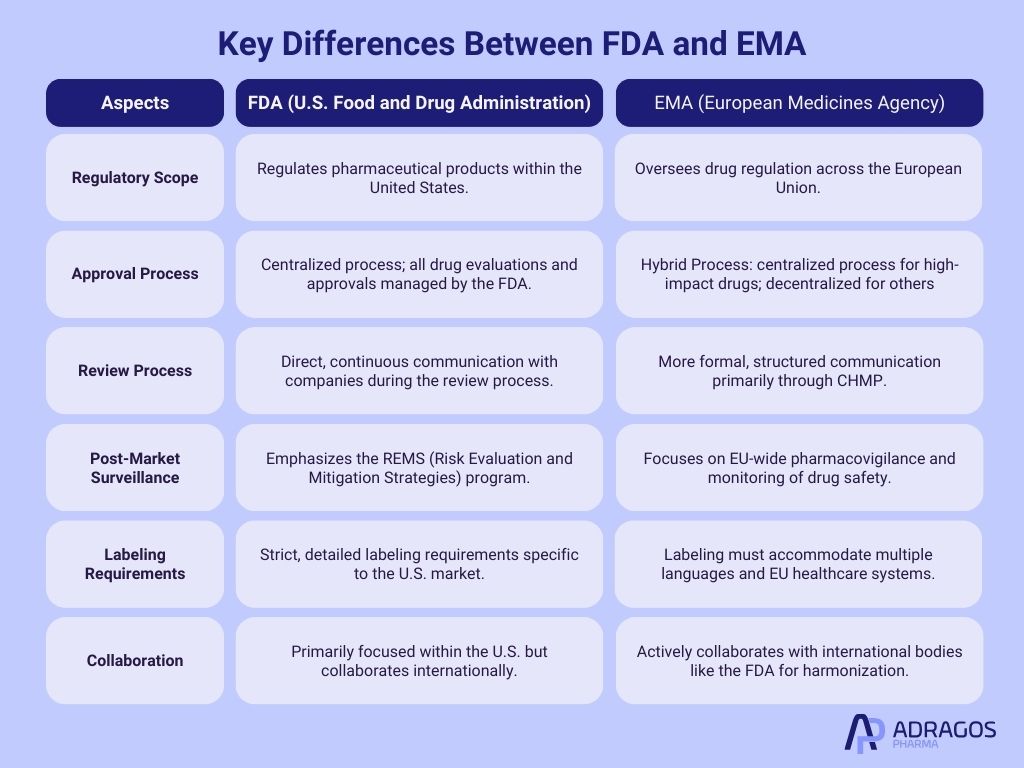
Principales différences entre la FDA et l’EMA
Bien qu’il y ait une tendance croissante à l’harmonisation, des différences significatives subsistent dans les approches réglementaires entre les régions. La FDA et l’EMA sont toutes deux des organismes de réglementation essentiels responsables d’assurer la sécurité, l’efficacité et la qualité des produits pharmaceutiques, cependant elles opèrent dans des cadres réglementaires différents et ont des processus distincts.
La FDA, en tant qu’autorité de réglementation aux États-Unis, suit une approche plus centralisée, où toutes les approbations et réglementations de médicaments sont gérées directement par l’agence. En revanche, l’EMA, qui sert l’Union européenne, coordonne avec les agences réglementaires nationales à travers les États membres de l’UE, utilisant un processus centralisé pour les médicaments à fort impact, tandis qu’un processus décentralisé pour d’autres, impliquant les agences nationales.
Une autre différence significative réside dans leurs processus d’examen respectifs : la FDA s’engage souvent dans une communication plus directe avec les entreprises pharmaceutiques pendant la phase d’examen, permettant un processus plus itératif, tandis que l’évaluation de l’EMA, menée par le Comité des médicaments à usage humain (CHMP), est généralement plus formalisée et structurée.
De plus, la FDA met fortement l’accent sur la surveillance post-commercialisation à travers son programme REMS (Stratégies d’Évaluation et d’Atténuation des Risques), tandis que l’EMA se concentre particulièrement sur la pharmacovigilance dans l’ensemble de l’UE.
Ces différences structurelles et procédurales influencent la manière dont les entreprises pharmaceutiques abordent le développement de médicaments, les soumissions réglementaires et les stratégies d’entrée sur le marché aux États-Unis et en Europe.
Réglementations pharmaceutiques internationales
Harmonisation par le biais du Conseil international
Le Conseil International d’Harmonisation (ICH) est un acteur clé dans le paysage réglementaire pharmaceutique mondial. L’ICH œuvre à harmoniser les exigences techniques pour le développement et l’approbation des médicaments dans différentes régions, réduisant ainsi la nécessité de tests en double et rationalisant le processus réglementaire. En adhérant aux directives de l’ICH, les entreprises pharmaceutiques peuvent s’assurer que leurs produits répondent aux normes nécessaires pour l’approbation sur de multiples marchés.
Conformité aux normes mondiales
La conformité aux normes mondiales est essentielle pour les entreprises pharmaceutiques qui souhaitent commercialiser leurs produits à l’échelle internationale. Cela inclut l’adhésion aux directives établies par l’ICH, ainsi que la conformité aux réglementations régionales aux États-Unis, en Europe et sur d’autres marchés. En se tenant informées des changements réglementaires et en travaillant en étroite collaboration avec les agences réglementaires, les entreprises pharmaceutiques peuvent s’assurer que leurs produits répondent aux normes nécessaires pour une distribution mondiale.
Défis réglementaires et solutions
Défis courants en matière de conformité réglementaire pharmaceutique
Les entreprises pharmaceutiques font face à plusieurs défis pour atteindre et maintenir la conformité réglementaire. Ces défis comprennent le suivi des réglementations en constante évolution, la gestion de la complexité des exigences réglementaires mondiales et la garantie que toute la documentation est précise et à jour. La non-conformité aux normes réglementaires peut entraîner des retards coûteux, des amendes et des dommages à la réputation d’une entreprise.
Stratégies pour surmonter les obstacles réglementaires
Pour surmonter les défis réglementaires, les entreprises pharmaceutiques doivent adopter une approche proactive de la conformité. Cela inclut de se tenir informé des changements réglementaires, d’investir dans la formation des employés et de travailler en étroite collaboration avec les agences réglementaires. En adoptant une approche proactive de la conformité réglementaire, les entreprises pharmaceutiques peuvent minimiser le risque de non-conformité et s’assurer que leurs produits répondent aux normes les plus élevées de sécurité et de qualité.
Les réglementations pharmaceutiques sont essentielles pour garantir que tous les médicaments sont sûrs, efficaces et de haute qualité. La conformité à ces réglementations est cruciale pour les entreprises pharmaceutiques, car elle protège non seulement la santé publique mais garantit également que les produits peuvent être mis sur le marché de manière efficiente. En se tenant informées des changements réglementaires et en travaillant en étroite collaboration avec les agences réglementaires, les entreprises pharmaceutiques peuvent naviguer dans le paysage réglementaire complexe et mettre sur le marché des médicaments sûrs et efficaces.
Questions fréquemment posées sur les réglementations pharmaceutiques
Quelles réglementations les entreprises pharmaceutiques suivent-elles ?
Les entreprises pharmaceutiques doivent suivre une large gamme de réglementations, y compris les directives CGMP, les réglementations de la FDA et les normes internationales établies par des organismes tels que l’ICH.
Que sont les directives 21 CFR dans l’industrie pharmaceutique ?
Les directives 21 CFR sont un ensemble de réglementations appliquées par la FDA qui couvrent divers aspects de la fabrication, de l’étiquetage et de la distribution des médicaments pour garantir la qualité et la sécurité des produits.
Quelle est la réglementation pharmaceutique aux États-Unis ?
Les réglementations pharmaceutiques aux États-Unis sont principalement régies par la FDA et les conseils de pharmacie au niveau des États, garantissant que tous les produits pharmaceutiques répondent aux normes de sécurité et d’efficacité.
Que sont les réglementations BPF ?
Les réglementations de Bonnes Pratiques de Fabrication (BPF) sont des directives qui garantissent que les médicaments sont constamment produits et contrôlés selon des normes de qualité.