
Navigating the pharmaceutical industry can be challenging and resource-intensive. The 505(b)(2) New Drug Application (NDA) offers a streamlined route within the drug approval process, allowing companies to bring innovative products to market more efficiently. This article delves into the 505(b)(2) pathway, its benefits, and its critical role in drug development, providing pharmaceutical companies with the information they need to optimize their strategies.
What is 505b2?
The 505(b)(2) new drug application (NDA) is one of three U.S. Food and Drug Administration (FDA) drug approval pathways, representing an appealing regulatory strategy for many clients. Established by the Hatch-Waxman Amendments of 1984, the 505(b)(2) pathway refers to a section of the Federal Food, Drug, and Cosmetic Act. In Europe, these kind of innovations have been commonly named after “Super generics” or “Generics+,” but lately, “Value Added Medicines” has become the go-to term.
The provisions of 505(b)(2) were created to help avoid unnecessary duplication of studies already performed on a previously approved drug, also known as the reference or listed drug. This pathway allows the FDA to rely on data not developed by the NDA applicant. A comprehensive development program is crucial for maximizing the streamlining capability of the 505(b)(2) approach and expediting the regulatory approval process.
A 505(b)(2) NDA contains full safety and effectiveness reports but allows at least some of the information required for NDA approval, such as safety and efficacy information on the active ingredient, to come from studies not conducted by or for the applicant. This can result in a much less expensive and much faster route to approval compared with a traditional development path, such as the 505(b)(1), while creating new, differentiated products with tremendous commercial value.
The Three Regulatory Pathways for Drug Approval
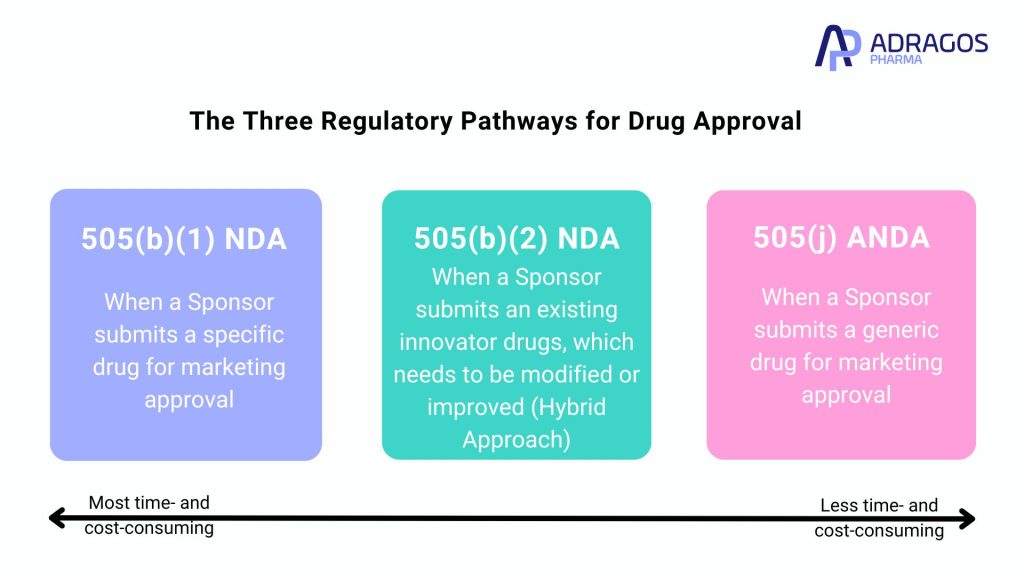
1. 505(b)(1) NDA
The 505(b)(1) NDA is a comprehensive application that relies entirely on original data. Every study conducted under this pathway is specifically tailored for the drug by the sponsor and serves as the foundational data for FDA approval. This pathway is the most resource-intensive and time-consuming of the three.
2. 505(j) ANDA
This pathway is designed for the approval of generic drugs. The 505(j) ANDA becomes relevant when a patented innovator drug is approaching its expiration. The central criterion for approval through this pathway is demonstrating bioequivalence to the innovator product. For oral dosage forms, a food effect study is typically required to ensure similar efficacy and safety profiles.
3. 505(b)(2) NDA
A hybrid application, the 505(b)(2) NDA combines aspects of both the full NDA and ANDA. This pathway is ideal for modified or improved versions of existing innovator drugs, leading to the creation of a distinct drug product with its own exclusivity rights. The process incorporates pre-existing data and new findings to facilitate a more efficient approval process. Preparing clinical trial materials that mirror the commercial manufacturing process for Phase 1 studies is crucial, involving significant CMC work before study initiation, including the preparation of stability batches for shelf-life determination. In Europe, a similar process is known as the Hybrid application, reflecting its combined nature.
Drug Candidates with 505b2 Potential
New Dosage Forms
A company may wish to create a new dosage form that is faster acting, combines two active ingredients in a novel way, or provides a route of administration or mechanism of drug delivery that patients or doctors prefer over previous versions.
New Indications
Companies might seek approval for a new indication for an already-approved drug or carry out an Rx-to-OTC switch. Such new products often contain well-understood active ingredients present in existing, approved drug products.
Benefits of the 505b2 Pathway
Lower Risk and Cost
The 505(b)(2) pathway is particularly valuable for pharmaceutical and generics companies looking to alleviate competitive forces in their environments while benefiting from a development process that eliminates most nonclinical studies and extensive safety and efficacy tests. The pathway offers a relatively lower risk due to previous drug approval, and the development process is typically faster and less expensive.
Market Exclusivity and Orphan Drug Exclusivity
The 505(b)(2) pathway also presents an opportunity for market exclusivity ranging from 3 to 7 years, providing a significant commercial advantage and allowing companies to recoup their investment before facing generic competition. Additionally, products approved under this pathway can qualify for various types of market exclusivity, including orphan drug exclusivity for seven years.
Commercial Viability
Ensuring commercial viability involves rigorous candidate assessment and strategic planning. Developers must evaluate market demand, potential competition, and strategically position their products for maximum impact.
Navigating the 505b2 Submission Process
Candidate Identification
Selecting appropriate drug products for the 505(b)(2) pathway is crucial. This step involves a few nonclinical studies to ensure that the new product’s pharmacokinetic (PK) profile is as favorable as the innovator product.
Candidate Assessment
Rigorous evaluation of the candidate’s scientific, medical, regulatory, and commercial viability is essential. This process helps validate the product concept and minimizes costly mistakes.
Product Planning
Strategic incorporation of existing data during product planning is vital. Developers should evaluate potential market exclusivity opportunities and plan their development programs accordingly.
Pre-IND Meeting
The pre-IND meeting with the FDA is a pivotal step in the 505(b)(2) pathway. This meeting aims to secure FDA input on planned studies and strategies, streamlining the approval process and reducing costs.
Detailed Steps in the 505b2 Approval Process
Conducting Bridging Studies
Some differences in formulation or administration might require additional clinical studies, but these are typically less extensive than those required for original innovator drugs. Often, a bioequivalence study suffices to demonstrate similarity.
Incorporating Pre-Existing Data
The ability to use pre-existing data significantly accelerates the approval process. By referencing the safety and efficacy data of the original drug, applicants can bypass the need for extensive new studies.
Strategic Use of Existing Data
Incorporating existing data into the development strategy is essential for optimizing the approval process. Evaluating potential market exclusivity and planning development programs accordingly can enhance the product’s value proposition.
Overcoming Challenges
Regulatory Challenges
Navigating the 505(b)(2) pathway requires a thorough understanding of regulatory requirements. Companies must ensure compliance with FDA guidelines and effectively manage the approval process.
Market Considerations
Market considerations include assessing competition, evaluating market demand, and ensuring the product’s commercial viability. Strategic positioning is crucial for maximizing market impact and profitability.
The 505(b)(2) pathway offers pharmaceutical companies a valuable opportunity to bring innovative products to market more efficiently and cost-effectively. By leveraging pre-existing data and minimizing redundant studies, this pathway facilitates a streamlined approval process, reducing time and financial investment.
FAQs about 505b2
What is the difference between NDA and 505(b)(2)?
An NDA (New Drug Application) relies entirely on original data, while a 505(b)(2) application allows the use of pre-existing data to streamline the approval process.
What is the difference between ANDA and 505(b)(2)?
ANDA (Abbreviated New Drug Application) is used for generic drugs and requires demonstrating bioequivalence to the innovator product. In contrast, a 505(b)(2) application is used for modified or improved versions of existing drugs and allows the use of pre-existing data.
What is a new molecular entity 505(b)(2)?
A new molecular entity 505(b)(2) refers to a new chemical entity developed through the 505(b)(2) pathway, leveraging pre-existing data to streamline the approval process.
What is the European equivalent of 505(b)(2)?
The European equivalent of the 505(b)(2) pathway is known as the Hybrid application, based on Article 10 of Directive 2001/83/EC. This pathway, similar to 505(b)(2), allows for the use of existing data from a reference medicinal product, supplemented by new clinical or pre-clinical data to support changes in formulation, dosage form, strength, or indication.
What is the probability of a 505(b)(2) being successful?
The probability of success for a 505(b)(2) application is generally higher than that of a traditional 505(b)(1) NDA due to the reliance on existing data, which reduces the risk and uncertainty associated with drug development. While exact success rates can vary based on the specific drug and modifications being proposed, industry analyses suggest that 505(b)(2) applications have a higher likelihood of approval, often cited to be around 70-75%, compared to the significantly lower success rates typically associated with new molecular entity (NME) approvals.